2. How-to¶
This notebook serves as a practical guide to common questions users might have.
Table of content
[1]:
import MDAnalysis as mda
import prolif as plf
import pandas as pd
import numpy as np
[2]:
u = mda.Universe(plf.datafiles.TOP, plf.datafiles.TRAJ)
lig = u.select_atoms("resname LIG")
prot = u.select_atoms("protein")
lmol = plf.Molecule.from_mda(lig)
pmol = plf.Molecule.from_mda(prot)
2.1. Changing the parameters for an interaction¶
You can list all the available interactions as follow:
[3]:
plf.Fingerprint.list_available(show_hidden=True)
[3]:
['Anionic',
'CationPi',
'Cationic',
'EdgeToFace',
'FaceToFace',
'HBAcceptor',
'HBDonor',
'Hydrophobic',
'Interaction',
'MetalAcceptor',
'MetalDonor',
'PiCation',
'PiStacking',
'XBAcceptor',
'XBDonor',
'_BaseCationPi',
'_BaseHBond',
'_BaseIonic',
'_BaseMetallic',
'_BaseXBond',
'_Distance']
In this example, we’ll redefine the hydrophobic interaction with a shorter distance.
You have the choice between overwriting the original hydrophobic interaction with the new one, or giving it an original name.
Let’s start with a test case: with the default parameters, Y109 is interacting with our ligand.
[4]:
fp = plf.Fingerprint()
fp.hydrophobic(lmol, pmol["TYR109.A"])
[4]:
True
2.1.1. Overwriting the original interaction¶
You have to define a class that inherits one of the classes listed in the prolif.interactions module.
[5]:
class Hydrophobic(plf.interactions.Hydrophobic):
def __init__(self):
super().__init__(distance=4.0)
/home/docs/checkouts/readthedocs.org/user_builds/prolif/conda/v0.3.2/lib/python3.9/site-packages/prolif/interactions.py:55: UserWarning: The 'Hydrophobic' interaction has been superseded by a new class with id 0x55c9a771a5f0
warnings.warn(f"The {name!r} interaction has been superseded by a "
[6]:
fp = plf.Fingerprint()
fp.hydrophobic(lmol, pmol["TYR109.A"])
[6]:
False
The interaction is not detected anymore. You can reset to the default interaction like so:
[7]:
class Hydrophobic(plf.interactions.Hydrophobic):
pass
fp = plf.Fingerprint()
fp.hydrophobic(lmol, pmol["TYR109.A"])
/home/docs/checkouts/readthedocs.org/user_builds/prolif/conda/v0.3.2/lib/python3.9/site-packages/prolif/interactions.py:55: UserWarning: The 'Hydrophobic' interaction has been superseded by a new class with id 0x55c9a72a8db0
warnings.warn(f"The {name!r} interaction has been superseded by a "
[7]:
True
2.1.2. Reparameterizing an interaction with another name¶
The steps are identical to above, just give the class a different name:
[8]:
class CustomHydrophobic(plf.interactions.Hydrophobic):
def __init__(self):
super().__init__(distance=4.0)
fp = plf.Fingerprint()
fp.hydrophobic(lmol, pmol["TYR109.A"])
[8]:
True
[9]:
fp.customhydrophobic(lmol, pmol["TYR109.A"])
[9]:
False
[10]:
fp = plf.Fingerprint(["Hydrophobic", "CustomHydrophobic"])
fp.bitvector(lmol, pmol["TYR109.A"])
[10]:
array([ True, False])
2.2. Writing your own interaction¶
Before you dive into this section, make sure that there isn’t already an interaction that could just be reparameterized to do what you want!
For this, the best is to check the section of the documentation corresponding to the prolif.interactions module. There are some generic interactions, like the _Distance class, if you just need to define two chemical moieties within a certain distance. Both the Hydrophobic, Ionic, and Metallic interactions inherit from this class!
With that being said, there are a few rules that you must respect when writing your own interaction:
Inherit the ProLIF Interaction class
This class is located in prolif.interactions.Interaction. If for any reason you must inherit from another class, you can also define the prolif.interactions._InteractionMeta as a metaclass.
Naming convention
Your class name must not start with _ or be named Interaction. For non-symmetrical interactions, like hydrogen bonds or salt-bridges, the convention used here is to named the class after the function of the ligand. For example, the class HBDonor detects if a ligand acts as a hydrogen bond donor, and the class Cationic detects if a ligand acts as a cation.
Define a ``detect`` method
This method takes exactly two positional arguments (and as many named arguments as you need): a ligand Residue or Molecule and a protein Residue or Molecule (in this order).
Return value(s) for the ``detect`` method
There are two possibilities here, depending on whether or not you want to access the indices of atoms responsible for the interaction. If you don’t need this information, just return True if the interaction is detected, False otherwise. If you need to access atomic indices, you must return the following items in this order:
TrueorFalsefor the detection of the interactionThe index of the ligand atom, or None if not detected
The index of the protein atom, or None if not detected
[11]:
from scipy.spatial import distance_matrix
# without atom indices
class CloseContact(plf.interactions.Interaction):
def detect(self, res1, res2, threshold=2.0):
dist_matrix = distance_matrix(res1.xyz, res2.xyz)
if (dist_matrix <= threshold).any():
return True
return False
fp = plf.Fingerprint()
fp.closecontact(lmol, pmol["ASP129.A"])
[11]:
True
[12]:
# with atom indices
class CloseContact(plf.interactions.Interaction):
def detect(self, res1, res2, threshold=2.0):
dist_matrix = distance_matrix(res1.xyz, res2.xyz)
mask = dist_matrix <= threshold
if mask.any():
res1_i, res2_i = np.where(mask)
# return the first solution
return True, res1_i[0], res2_i[0]
return False, None, None
fp = plf.Fingerprint()
fp.closecontact(lmol, pmol["ASP129.A"])
/home/docs/checkouts/readthedocs.org/user_builds/prolif/conda/v0.3.2/lib/python3.9/site-packages/prolif/interactions.py:55: UserWarning: The 'CloseContact' interaction has been superseded by a new class with id 0x55c9a6d885c0
warnings.warn(f"The {name!r} interaction has been superseded by a "
[12]:
True
By default, the fingerprint will modify all interaction classes to only return the boolean value. To get the atom indices you must choose one of the following options:
Call
fp.to_dataframe(return_atoms=True)
Use the
return_atoms=Trueargument when calling thegeneratemethod:
[13]:
ifp = fp.generate(lmol, pmol, return_atoms=True)
# check the interactino between the ligand and ASP129
ifp[(plf.ResidueId("LIG", 1, "G"),
plf.ResidueId("ASP", 129, "A"))]
[13]:
(array([ True, True, False, True, False, False, False, False]),
[8, 52, None, 13, None, None, None, None],
[7, 8, None, 8, None, None, None, None])
Use the
__wrapped__argument when calling the class as a fingerprint method:
[14]:
fp.closecontact.__wrapped__(lmol, pmol["ASP129.A"])
[14]:
(True, 52, 8)
Use the
bitvector_atomsmethod instead ofbitvector:
[15]:
fp = plf.Fingerprint(["CloseContact"])
bv, lig_ix, prot_ix = fp.bitvector_atoms(lmol, pmol["ASP129.A"])
bv, lig_ix, prot_ix
[15]:
(array([ True]), [52], [8])
Directly use your class:
[16]:
cc = CloseContact()
cc.detect(lmol, pmol["ASP129.A"])
[16]:
(True, 52, 8)
2.3. Working with docking poses instead of MD simulations¶
ProLIF currently provides file readers for MOL2, SDF and PDBQT files. The API is slightly different compared to the quickstart example but the end result is the same.
Please note that this part of the tutorial is only suitable for interactions between one protein and several ligands, or in more general terms, between one molecule with multiple residues and one molecule with a single residue. This is not suitable for protein-protein or DNA-protein interactions.
Let’s start by loading the protein. Here I’m using a PDB file but you can use any format supported by MDAnalysis as long as it contains explicit hydrogens.
[17]:
# load protein
prot = mda.Universe(plf.datafiles.datapath / "vina" / "rec.pdb")
prot = plf.Molecule.from_mda(prot)
prot.n_residues
[17]:
302
2.3.1. Using an SDF file¶
[18]:
# load ligands
path = str(plf.datafiles.datapath / "vina" / "vina_output.sdf")
lig_suppl = list(plf.sdf_supplier(path))
# generate fingerprint
fp = plf.Fingerprint()
fp.run_from_iterable(lig_suppl, prot)
df = fp.to_dataframe()
df
[18]:
| ligand | UNL1 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| protein | TYR38.A | TYR40.A | SER106.A | TYR109.A | CYS122.A | ASP123.A | TRP125.A | ... | PRO338.B | PHE346.B | HSE347.B | LEU348.B | PHE351.B | ASP352.B | THR355.B | TYR359.B | |||||
| interaction | Hydrophobic | HBAcceptor | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | ... | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking |
| Frame | |||||||||||||||||||||
| 0 | False | False | False | False | True | False | False | False | False | False | ... | False | False | False | False | True | True | True | True | False | False |
| 1 | False | False | False | False | True | False | False | False | True | False | ... | False | False | False | False | True | True | True | True | False | False |
| 2 | False | False | False | False | True | False | False | False | True | False | ... | False | False | False | False | True | False | True | True | False | False |
| 3 | True | False | False | False | False | False | False | False | False | False | ... | False | False | False | False | True | False | True | True | True | False |
| 4 | True | True | False | False | True | False | False | False | False | False | ... | True | True | True | True | True | True | True | False | False | False |
| 5 | False | False | False | False | True | False | False | False | True | False | ... | False | False | False | False | True | False | True | True | False | False |
| 6 | True | False | True | True | True | True | False | False | True | True | ... | False | False | False | True | True | True | False | True | True | True |
| 7 | True | False | False | False | True | False | False | False | True | False | ... | True | False | True | True | True | True | False | True | False | False |
| 8 | False | False | False | False | False | False | True | True | False | False | ... | False | False | False | False | False | False | False | False | False | False |
9 rows × 47 columns
Please note that converting the lig_suppl to a list is optionnal (and maybe not suitable for large files) as it will load all the ligands in memory, but it’s nicer to track the progression with the progress bar.
If you want to calculate the Tanimoto similarity between your docked poses and a reference ligand, here’s how to do it.
We first need to generate the interaction fingerprint for the reference, and concatenate it to the previous one
[19]:
# load the reference
ref = mda.Universe(plf.datafiles.datapath / "vina" / "lig.pdb")
ref = plf.Molecule.from_mda(ref)
# generate IFP
fp.run_from_iterable([ref], prot)
df0 = fp.to_dataframe()
df0.rename({0: "ref"}, inplace=True)
# drop the ligand level on both dataframes
df0.columns = df0.columns.droplevel(0)
df.columns = df.columns.droplevel(0)
# concatenate them
df = (pd.concat([df0, df])
.fillna(False)
.sort_index(axis=1, level=0,
key=lambda index: [plf.ResidueId.from_string(x) for x in index]))
df
[19]:
| protein | TYR38.A | TYR40.A | SER106.A | TYR109.A | CYS122.A | ASP123.A | TRP125.A | ... | PRO338.B | PHE346.B | HSE347.B | LEU348.B | PHE351.B | ASP352.B | THR355.B | TYR359.B | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| interaction | HBAcceptor | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | ... | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking |
| Frame | |||||||||||||||||||||
| ref | False | False | False | False | True | False | False | False | True | False | ... | True | False | False | False | True | False | True | True | True | False |
| 0 | False | False | False | False | True | False | False | False | False | False | ... | False | False | False | False | True | True | True | True | False | False |
| 1 | False | False | False | False | True | False | False | False | True | False | ... | False | False | False | False | True | True | True | True | False | False |
| 2 | False | False | False | False | True | False | False | False | True | False | ... | False | False | False | False | True | False | True | True | False | False |
| 3 | False | True | False | False | False | False | False | False | False | False | ... | False | False | False | False | True | False | True | True | True | False |
| 4 | True | True | False | False | True | False | False | False | False | False | ... | True | True | True | True | True | True | True | False | False | False |
| 5 | False | False | False | False | True | False | False | False | True | False | ... | False | False | False | False | True | False | True | True | False | False |
| 6 | False | True | True | True | True | True | False | False | True | True | ... | False | False | False | True | True | True | False | True | True | True |
| 7 | False | True | False | False | True | False | False | False | True | False | ... | True | False | True | True | True | True | False | True | False | False |
| 8 | False | False | False | False | False | False | True | True | False | False | ... | False | False | False | False | False | False | False | False | False | False |
10 rows × 50 columns
Lastly, we can convert the dataframe to a list of RDKit bitvectors to finally compute the Tanimoto similarity between our reference pose and the docking poses generated by Vina:
[20]:
from rdkit import DataStructs
bvs = plf.to_bitvectors(df)
for i, bv in enumerate(bvs[1:]):
tc = DataStructs.TanimotoSimilarity(bvs[0], bv)
print(f"{i}: {tc:.3f}")
0: 0.633
1: 0.455
2: 0.484
3: 0.433
4: 0.286
5: 0.690
6: 0.278
7: 0.469
8: 0.297
Interestingly, the best scored docking pose (#0) isn’t the most similar to the reference (#5)
2.3.2. Using a MOL2 file¶
The input mol2 file can contain multiple ligands in different conformations.
[21]:
# load ligands
path = plf.datafiles.datapath / "vina" / "vina_output.mol2"
lig_suppl = list(plf.mol2_supplier(path))
# generate fingerprint
fp = plf.Fingerprint()
fp.run_from_iterable(lig_suppl, prot)
df = fp.to_dataframe()
df
[21]:
| ligand | UNL1 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| protein | TYR38.A | TYR40.A | SER106.A | TYR109.A | CYS122.A | ASP123.A | TRP125.A | ... | PRO338.B | PHE346.B | HSE347.B | LEU348.B | PHE351.B | ASP352.B | THR355.B | TYR359.B | |||||
| interaction | Hydrophobic | HBAcceptor | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | ... | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking |
| Frame | |||||||||||||||||||||
| 0 | False | False | False | False | True | False | False | False | False | False | ... | False | False | False | False | True | True | True | True | False | False |
| 1 | False | False | False | False | True | False | False | False | True | False | ... | False | False | False | False | True | True | True | True | False | False |
| 2 | False | False | False | False | True | False | False | False | True | False | ... | False | False | False | False | True | False | True | True | False | False |
| 3 | True | False | False | False | False | False | False | False | False | False | ... | False | False | False | False | True | False | True | True | True | False |
| 4 | True | True | False | False | True | False | False | False | False | False | ... | True | True | True | True | True | True | True | False | False | False |
| 5 | False | False | False | False | True | False | False | False | True | False | ... | False | False | False | False | True | False | True | True | False | False |
| 6 | True | False | True | True | True | True | False | False | True | True | ... | False | False | False | True | True | True | False | True | True | True |
| 7 | True | False | False | False | True | False | False | False | True | False | ... | True | False | True | True | True | True | False | True | False | False |
| 8 | False | False | False | False | False | False | True | True | False | False | ... | False | False | False | False | False | False | False | False | False | False |
9 rows × 47 columns
2.3.3. Using PDBQT files¶
The typical use case here is getting the IFP from AutoDock Vina’s output. It requires a few additional steps and informations compared to other formats like MOL2, since the PDBQT format gets rid of most hydrogen atoms and doesn’t contain bond order information.
The prerequisites for a successfull usage of ProLIF in this case is having external files that contain bond orders and formal charges for your ligand (like SMILES, SDF or MOL2), or at least a file with explicit hydrogen atoms.
Please note that your PDBQT input must have a single model per file (this is required by MDAnalysis). Splitting a multi-model file can be done using the vina_split command-line tool that comes with AutoDock Vina: vina_split --input vina_output.pdbqt
Let’s start by loading our “template” file with bond orders. It can be a SMILES string, MOL2, SDF file or anything supported by RDKit.
[22]:
from rdkit import Chem
from rdkit.Chem import AllChem
template = Chem.MolFromSmiles("C[NH+]1CC(C(=O)NC2(C)OC3(O)C4CCCN4C(=O)C"
"(Cc4ccccc4)N3C2=O)C=C2c3cccc4[nH]cc(c34)CC21")
template
[22]:
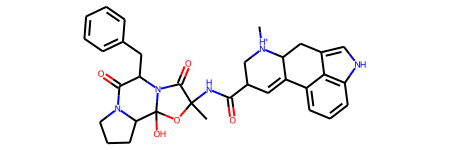
Next, we’ll use the PDBQT supplier which loads each file from a list of paths, and assigns bond orders and charges using the template. The template and PDBQT file must have the exact same atoms, even hydrogens, otherwise no match will be found. Since PDBQT files partially keep the hydrogen atoms, we have the choice between:
Manually selecting where to add the hydrogens on the template, do the matching, then add the remaining hydrogens (not covered here)
Or just remove the hydrogens from the PDBQT file, do the matching, then add all hydrogens.
This last option will delete the coordinates of your hydrogens atoms and replace them by the ones generated by RDKit, but unless you’re working with an exotic system this should be fine.
For the protein, there’s usually no need to load the PDBQT that was used by Vina. The original file that was used to generate the PDBQT can be used directly, but it must contain explicit hydrogen atoms:
[23]:
# load ligands
pdbqt_files = sorted(plf.datafiles.datapath.glob("vina/*.pdbqt"))
lig_suppl = list(plf.pdbqt_supplier(pdbqt_files, template))
# generate fingerprint
fp = plf.Fingerprint()
fp.run_from_iterable(lig_suppl, prot)
df = fp.to_dataframe()
df
/home/docs/checkouts/readthedocs.org/user_builds/prolif/conda/v0.3.2/lib/python3.9/site-packages/MDAnalysis/topology/guessers.py:146: UserWarning: Failed to guess the mass for the following atom types: A
warnings.warn("Failed to guess the mass for the following atom types: {}".format(atom_type))
/home/docs/checkouts/readthedocs.org/user_builds/prolif/conda/v0.3.2/lib/python3.9/site-packages/MDAnalysis/topology/guessers.py:146: UserWarning: Failed to guess the mass for the following atom types: HD
warnings.warn("Failed to guess the mass for the following atom types: {}".format(atom_type))
/home/docs/checkouts/readthedocs.org/user_builds/prolif/conda/v0.3.2/lib/python3.9/site-packages/MDAnalysis/topology/guessers.py:146: UserWarning: Failed to guess the mass for the following atom types: OA
warnings.warn("Failed to guess the mass for the following atom types: {}".format(atom_type))
/home/docs/checkouts/readthedocs.org/user_builds/prolif/conda/v0.3.2/lib/python3.9/site-packages/MDAnalysis/converters/RDKit.py:421: UserWarning: No `bonds` attribute in this AtomGroup. Guessing bonds based on atoms coordinates
warnings.warn(
RDKit ERROR: [11:36:29] Explicit valence for atom # 21 N, 4, is greater than permitted
/home/docs/checkouts/readthedocs.org/user_builds/prolif/conda/v0.3.2/lib/python3.9/site-packages/MDAnalysis/converters/RDKit.py:448: UserWarning: Could not sanitize molecule: failed during step rdkit.Chem.rdmolops.SanitizeFlags.SANITIZE_PROPERTIES
warnings.warn("Could not sanitize molecule: "
RDKit ERROR: [11:36:29] Explicit valence for atom # 21 N, 4, is greater than permitted
RDKit ERROR: [11:36:29] Explicit valence for atom # 21 N, 4, is greater than permitted
RDKit ERROR: [11:36:29] Explicit valence for atom # 21 N, 4, is greater than permitted
RDKit ERROR: [11:36:29] Explicit valence for atom # 21 N, 4, is greater than permitted
RDKit ERROR: [11:36:30] Explicit valence for atom # 21 N, 4, is greater than permitted
RDKit ERROR: [11:36:30] Explicit valence for atom # 21 N, 4, is greater than permitted
RDKit ERROR: [11:36:30] Explicit valence for atom # 21 N, 4, is greater than permitted
RDKit ERROR: [11:36:30] Explicit valence for atom # 21 N, 4, is greater than permitted
[23]:
| ligand | LIG1.G | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| protein | TYR38.A | TYR40.A | SER106.A | TYR109.A | CYS122.A | ASP123.A | TRP125.A | ... | PRO338.B | PHE346.B | HSE347.B | LEU348.B | PHE351.B | ASP352.B | THR355.B | TYR359.B | |||||
| interaction | Hydrophobic | HBAcceptor | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | ... | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking |
| Frame | |||||||||||||||||||||
| 0 | False | False | False | False | True | False | False | False | False | False | ... | False | False | False | False | True | True | True | True | False | False |
| 1 | False | False | False | False | True | False | False | False | True | False | ... | False | False | False | False | True | True | True | True | False | False |
| 2 | False | False | False | False | True | False | False | False | True | False | ... | False | False | False | False | True | False | True | True | False | False |
| 3 | True | False | False | False | False | False | False | False | False | False | ... | False | False | False | False | True | False | True | True | True | False |
| 4 | True | True | False | False | True | False | False | False | False | False | ... | True | True | True | True | True | True | True | False | False | False |
| 5 | False | False | False | False | True | False | False | False | True | False | ... | False | False | False | False | True | False | True | True | False | False |
| 6 | True | False | True | True | True | True | False | False | True | True | ... | False | False | False | True | True | True | False | True | True | True |
| 7 | True | False | False | False | True | False | False | False | True | False | ... | True | False | True | True | True | True | False | True | False | False |
| 8 | False | False | False | False | False | False | True | True | False | False | ... | False | False | False | False | False | False | False | False | False | False |
9 rows × 47 columns